
2017 , Vol. 11 >Issue 02: 97 - 101
DOI: https://doi.org/10.3877/cma.j.issn.1674-0807.2017.02.008
乳腺癌血管生成拟态的分子机制研究进展
Copy editor: 宗贝歌
收稿日期: 2016-05-11
网络出版日期: 2024-12-04
基金资助
江苏省中医药管理局中医药科技创新基金项目(HZ0816KY)无锡市科技局项目(CSE31N1330)江南大学大学生创新创业训练计划项目(2016390Y)江南大学无锡医学院本科教育教学改革研究项目(JG2016YY008)
版权
Molecular mechanism of angiogenesis mimicry in breast cancer
Received date: 2016-05-11
Online published: 2024-12-04
Copyright
陈宇潇 , 倪成铭 , 张金梦 , 孙瑞凤 , 陈婷 , 张治宣 , 宫海凤 , 杨薇 , 赵涵 , 蔡维维 , 邱丽颖 , 冯磊 . 乳腺癌血管生成拟态的分子机制研究进展[J]. 中华乳腺病杂志(电子版), 2017 , 11(02) : 97 -101 . DOI: 10.3877/cma.j.issn.1674-0807.2017.02.008
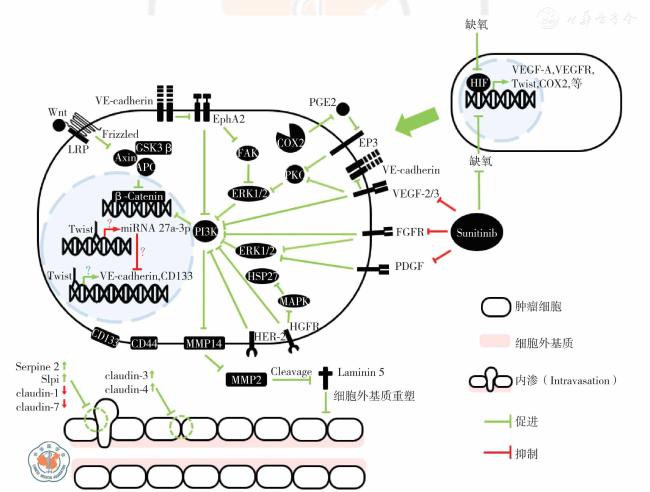
图1 乳腺癌血管生成拟态形成的分子机制示意图注:β-catenin 为β-连环蛋白;Axin 为轴抑制蛋白;APC 为肠腺瘤息肉病基因;COX 为环氧化酶;Claudin 为紧密连接蛋白;EGFR 为表皮生长因子受体;EP 为前列腺素类受体;EphA2 为促红细胞生成素产生肝细胞受A2;ERK 为细胞外信号调节激酶;Frizzled 为卷曲蛋白;FAK 为黏着斑激酶;FGFR 为成纤维生长因子受体;GSK3β 为糖原合成酶激酶3β;HER 为人类表皮生长因子受体;HSP 为热休克蛋白;HIF 为缺氧诱导因子;LRP 为低密度脂蛋白受体相关蛋白;Laminin 为层黏连蛋白;MMP 为基质金属蛋白酶;MAPK 为丝裂原激活的蛋白激酶;PDGF 为血小板源生长因子;PI3K 为磷脂酰肌醇-3-羟激酶;PG 为前列腺素;Serpine 为丝氨酸蛋白酶抑制剂;Slpi 为分泌性白细胞蛋白酶抑制因子;Sunitinib 为舒尼替尼;Twist 为上皮间质转化因子;VE-cadherin为血管内皮细胞钙黏蛋白;VEGF 为血管内皮生长因子;VEGFR 为血管内皮生长因子受体 |
| [1] |
Maniotis AJ, Folberg R, Hess A, et al. Vascular channel formation by human melanoma cells in vivo and in vitro: vasculogenic mimicry[J].Am J Pathol,1999,155(3):739-752.
|
| [2] |
Ricci-Vitiani L,Pallini R,Biffoni M,et al. Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells[J]. Nature,2010,468(7325):824-828.
|
| [3] |
Zhang D, Sun B, Zhao X, et al. Twist1 expression induced by sunitinib accelerates tumor cell vasculogenic mimicry by increasing the population of CD133+ cells in triple-negative breast cancer[J]. Mol Cancer,2014,13:207.
|
| [4] |
Liu T, Sun B, Zhao X, et al. OCT4 expression and vasculogenic mimicry formation positively correlate with poor prognosis in human breast cancer[J]. Int J Mol Sci,2014,15(11):19 634-19 649.
|
| [5] |
Wagenblast E, Soto M, Gutiérrez-ángel S, et al. A model of breast cancer heterogeneity reveals vascular mimicry as a driver of metastasis[J]. Nature,2015,520(7547):358-362.
|
| [6] |
Partridge AH, Rumble RB, Carey LA, et al. Chemotherapy and targeted therapy for women with human epidermal growth factor receptor 2-negative (or unknown) advanced breast cancer: American Society of Clinical Oncology Clinical Practice Guideline[J]. J Clin Oncol,2014,32(29):3307-3329.
|
| [7] |
Barrios CH, Liu MC, Lee SC, et al. Phase Ⅲrandomized trial of sunitinib versus capecitabine in patients with previously treated HER2-negative advanced breast cancer[J]. Breast Cancer Res Treat, 2010,121(1):121-131.
|
| [8] |
Crown JP, Diéras V, Staroslawska E, et al. Phase Ⅲtrial of sunitinib in combination with capecitabine versus capecitabine monotherapy for the treatment of patients with pretreated metastatic breast cancer[J]. J Clin Oncol,2013,31(23):2870-2878.
|
| [9] |
Kerbel RS. Reappraising antiangiogenic therapy for breast cancer[J].Breast,2011,20 Suppl 3:S56-60.
|
| [10] |
Yao N, Ren K, Jiang C, et al. Combretastatin A4 phosphate treatment induces vasculogenic mimicry formation of W256 breast carcinoma tumor in vitro and in vivo [J]. Tumour Biol, 2015, 36 (11):8499-8510.
|
| [11] |
Hess AR, Seftor EA, Gruman LM, et al. VE-cadherin regulates EphA2 in aggressive melanoma cells through a novel signaling pathway:implications for vasculogenic mimicry[J]. Cancer Biol Ther, 2006, 5(2):228-233.
|
| [12] |
Hess AR, Hendrix MJ. Focal adhesion kinase signaling and the aggressive melanoma phenotype [J]. Cell Cycle, 2006, 5 (5):478-480.
|
| [13] |
Hess AR, Seftor EA, Seftor RE, et al. Phosphoinositide 3-kinase regulates membrane Type 1-matrix metalloproteinase (MMP) and MMP-2 activity during melanoma cell vasculogenic mimicry[J]. Cancer Res,2003,63(16):4757-4762.
|
| [14] |
Hess AR, Postovit LM, Margaryan NV, et al. Focal adhesion kinase promotes the aggressive melanoma phenotype[J]. Cancer Res, 2005,65(21):9851-9860.
|
| [15] |
Seftor RE, Seftor EA, Koshikawa N, et al. Cooperative interactions of laminin 5 gamma2 chain, matrix metalloproteinase-2, and membrane type-1-matrix/metalloproteinase are required for mimicry of embryonic vasculogenesis by aggressive melanoma [J]. Cancer Res, 2001,61(17):6322-6327.
|
| [16] |
Sun MG, Shi JF, Li XY, et al. Targeting epirubicin plus quinacrine liposomes modified with DSPE-PEG2000-C (RGDfK) conjugate for eliminating invasive breast cancer[J]. J Biomed Nanotechnol, 2015,11(8):1339-1353.
|
| [17] |
Zeng F, Ju RJ, Liu L, et al. Application of functional vincristine plus dasatinib liposomes to deletion of vasculogenic mimicry channels in triple-negative breast cancer [ J]. Oncotarget, 2015, 6 (34):36 625-36 642.
|
| [18] |
Ju RJ, Li XT, Shi JF, et al. Liposomes, modified with PTD(HIV-1)peptide, containing epirubicin and celecoxib, to target vasculogenic mimicry channels in invasive breast cancer[J]. Biomaterials, 2014,35(26):7610-7621.
|
| [19] |
Shi JF, Sun MG, Li XY, et al. A combination of targeted sunitinib liposomes and targeted vinorelbine liposomes for treating invasive breast cancer[J]. J Biomed Nanotechnol,2015,11(9):1568-1582.
|
| [20] |
Sun T, Zhao N,Zhao XL,et al. Expression and functional significance of Twist1 in hepatocellular carcinoma: its role in vasculogenic mimicry[J]. Hepatology,2010,51(2):545-556.
|
| [21] |
Zhao N, Sun H,Sun B,et al. miR-27a-3p suppresses tumor metastasis and VM by down-regulating VE-cadherin expression and inhibiting EMT: an essential role for Twist-1 in HCC[J]. Sci Rep, 2016, 6:23 091.
|
| [22] |
Ristimäki A, Sivula A, Lundin J,et al. Prognostic significance of elevated cyclooxygenase-2 expression in breast cancer[J]. Cancer Res,2002,62(3):632-635.
|
| [23] |
Mosalpuria K, Hall C, Krishnamurthy S, et al. Cyclooxygenase-2 expression in non-metastatic triple-negative breast cancer patients[J].Mol Clin Oncol,2014,2(5):845-850.
|
| [24] |
Basu GD, Liang WS, Stephan DA, et al. A novel role for cyclooxygenase-2 in regulating vascular channel formation by human breast cancer cells[J]. Breast Cancer Res,2006,8(6):R69.
|
| [25] |
Robertson FM, Simeone AM, Lucci A, et al. Differential regulation of the aggressive phenotype of inflammatory breast cancer cells by prostanoid receptors EP3 and EP4 [J]. Cancer, 2010, 116 (11 Suppl):2806-2814.
|
| [26] |
Ravi M,Tentu S,Baskar G,et al. Molecular mechanism of anti-cancer activity of phycocyanin in triple-negative breast cancer cells[J]. BMC Cancer,2015,15:768.
|
| [27] |
Vartanian A, Stepanova E, Grigorieva I, et al. VEGFR1 and PKCalpha signaling control melanoma vasculogenic mimicry in a VEGFR2 kinase-independent manner [J]. Melanoma Res, 2011,21(2):91-98.
|
| [28] |
Spinella F, Caprara V, Di Castro V, et al. Endothelin-1 induces the transactivation of vascular endothelial growth factor receptor-3 and modulates cell migration and vasculogenic mimicry in melanoma cells[J]. J Mol Med (Berl),2013,91(3):395-405.
|
| [29] |
Karroum A,Mirshahi P,Faussat AM,et al. Tubular network formation by adriamycin-resistant MCF-7 breast cancer cells is closely linked to MMP-9 and VEGFR-2/VEGFR-3 over-expressions [ J]. Eur J Pharmacol,2012,685(1-3):1-7.
|
| [30] |
Gehmert S, Gehmert S, Prantl L, et al. Breast cancer cells attract the migration of adipose tissue-derived stem cells via the PDGF-BB/PDGFR-β signaling pathway[J]. Biochem Biophys Res Commun,2010,398(3):601-605.
|
| [31] |
Plantamura I, Casalini P, Dugnani E, et al. PDGFRβ and FGFR2 mediate endothelial cell differentiation capability of triple negative breast carcinoma cells[J]. Mol Oncol,2014,8(5):968-981.
|
| [32] |
Sun B, Zhang D, Zhao N, et al. Epithelial-to-endothelial transition and cancer stem cells: two cornerstones of vasculogenic mimicry in malignant tumors[J]. Oncotarget, 2016. doi: 10.18632/oncotarget.8461.
|
| [33] |
Fang X, Cai Y, Liu J, et al. Twist2 contributes to breast cancer progression by promoting an epithelial-mesenchymal transition and cancer stem-like cell self-renewal[J]. Oncogene, 2011, 30(47):4707-4720.
|
| [34] |
Sun T, Sun BC, Zhao XL, et al. Promotion of tumor cell metastasis and vasculogenic mimicry by way of transcription coactivation by Bcl-2 and Twist1: a study of hepatocellular carcinoma[J]. Hepatology,2011,54(5):1690-1706.
|
| [35] |
Luan YY, Liu ZM, Zhong JY, et al. Effect of grape seed proanthocyanidins on tumor vasculogenic mimicry in human triplenegative breast cancer cells[J]. Asian Pac J Cancer Prev, 2015,16(2):531-535.
|
| [36] |
Qi L,Song W,Liu Z,et al. Wnt3a promotes the vasculogenic mimicry formation of colon cancer via Wnt/β-Catenin signaling[J]. Int J Mol Sci,2015,16(8):18 564-18 579.
|
| [37] |
Medema JP. Cancer stem cells: the challenges ahead[J]. Nat Cell Biol,2013,15(4):338-344.
|
| [38] |
Liu TJ, Sun BC, Zhao XL, et al. CD133+ cells with cancer stem cell characteristics associates with vasculogenic mimicry in triple-negative breast cancer[J]. Oncogene,2013,32(5):544-553.
|
| [39] |
Paulis YW, Huijbers EJ, van der Schaft DW, et al. CD44 enhances tumor aggressiveness by promoting tumor cell plasticity [ J].Oncotarget,2015,6(23):19 634-19 646.
|
| [40] |
Liu T, Sun B, Zhao X, et al. USP44+ cancer stem cell subclones contribute to breast cancer aggressiveness by promoting vasculogenic mimicry[J]. Mol Cancer Ther,2015,14(9):2121-2131.
|
| [41] |
Lee CH, Wu YT, Hsieh HC, et al. Epidermal growth factor/heat shock protein 27 pathway regulates vasculogenic mimicry activity of breast cancer stem/progenitor cells [J]. Biochimie, 2014, 104:117-126.
|
| [42] |
van Malenstein H, Dekervel J, Verslype C, et al. Long-term exposure to sorafenib of liver cancer cells induces resistance with epithelial-tomesenchymal transition, increased invasion and risk of rebound growth[J]. Cancer Lett,2013,329(1):74-83.
|
| [43] |
Nagai T, Arao T, Furuta K, et al. Sorafenib inhibits the hepatocyte growth factor-mediated epithelial mesenchymal transition in hepatocellular carcinoma [J]. Mol Cancer Ther, 2011, 10 (1):169-177.
|
| [44] |
Liu T, Sun B, Zhao X, et al. HER2/neu expression correlates with vasculogenic mimicry in invasive breast carcinoma[J]. J Cell Mol Med,2013,17(1):116-122.
|
| [45] |
Cardone RA, Greco MR, Capulli M, et al. NHERF1 acts as a molecular switch to program metastatic behavior and organotropism via its PDZ domains[J]. Mol Biol Cell,2012,23(11):2028-2040.
|
| [46] |
Krämer F, White K, Kubbies M, et al. Genomic organization of claudin-1 and its assessment in hereditary and sporadic breast cancer[J]. Hum Genet,2000,107(3):249-256.
|
| [47] |
Kominsky SL, Argani P, Korz D, et al. Loss of the tight junction protein claudin-7 correlates with histological grade in both ductal carcinoma in situ and invasive ductal carcinoma of the breast[J].Oncogene,2003,22(13):2021-2033.
|
| [48] |
Harrell JC,Pfefferle AD,Zalles N,et al. Endothelial-like properties of claudin-low breast cancer cells promote tumor vascular permeability and metastasis[J]. Clin Exp Metastasis,2014,31(1):33-45.
|
| [49] |
Cui YF, Liu AH,An DZ,et al. Claudin-4 is required for vasculogenic mimicry formation in human breast cancer cells[J]. Oncotarget,2015,6(13):11 087-11 097.
|
/
| 〈 |
|
〉 |
{kind=link}
{kind=link}